简单t检验
在这一节中,我们将展示一下使用简单t检验和使用limma层次模型进行差异分析的区别。limma的相关参考文献已经列到参考资料中。
在这一节中,我们还民法了执行limma分析的基本步骤。需要注意的是,limma的功能非常强大,它有数百页的文档,我们不能详细地介绍这个包以及函数,有兴趣的同学可以直接查看它的文档。
spike-in数据集
我们先加载一批由Affymetrix生成的均一化(normalized)后的spike-in数据,如下所示:
|
|
简单来说,这里一共有16个mRNA的物种,它们都已经使用了固定的浓度,并混合起来使用了hgu95芯片进行了检验。这个子集中对于每个mRNA物种,ExpressionSet中的第一个三元组(trio)和第二个三元组有着固定的不同值。通过pData就能看到,如下所示:
|
|
如果要了解这批数据这样设计的原理,现在我们来看一下下面的四张spiked mRNA,原文如下:
To get a feel for the response of the array quantifications to this design, consider the following plots for four of the spiked mRNAs.
|
|
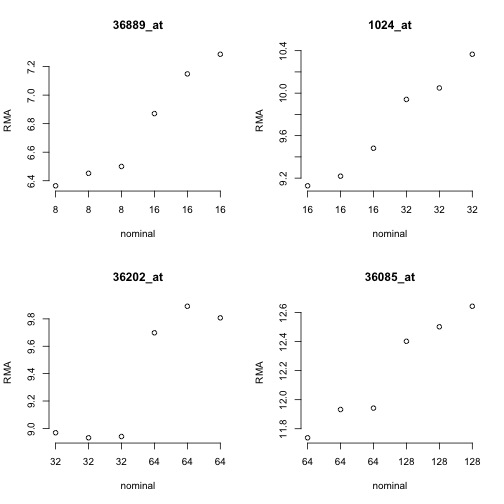
由于RMA(robust multi-array average,鲁棒多阵列平均 )是以log2转换为定量标准的,因此在归一化的检测值中观察到的差异是比较合理的。但是,在每个设计好的浓度内部,存在着相当大的变异。
rowttests
我们现在使用 genefilter 包中的 rowttests 函数来做一个简单的t检验,如下所示:
|
|
我们将根据p值,均值,以及特征值是否是spike-in值来设置颜色,如下所示:
|
|
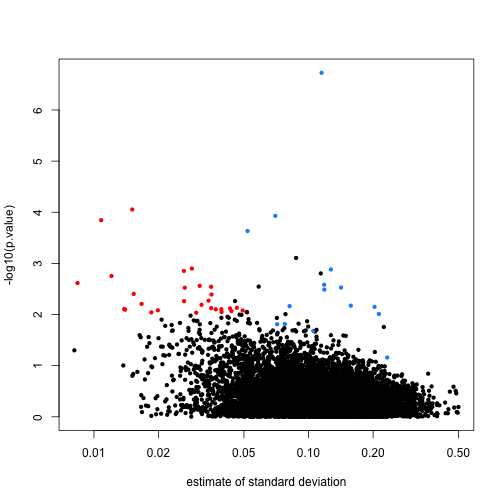
我们现在使用上面定义的颜色来绘制结果。我们将dm乘以-1,因为国我们感兴趣的是,第二组与第一组的差异(这是由lm与limma包默认情况下使用的差异)。spike-in基因是蓝色的,它们大多数有着较小的p值与较大的均值差异。红点表示较小p值的基因,同时也有着较小的均值差异。我们将会看到这些点使用limma后的变化,如下所示:
|
|

Note that the red genes have mostly low estimates of standard deviation.
|
|
limma步骤
以下是执行基本limma分析的三个步骤。我们指定coef=2,因为我感兴趣的是组与组之间的差异,而不是截距(intercept),如下所示:
|
|
topTable 会返回你定义的值排序的前几个基因。你还可以使用topTable通过指定none来要求返回所有的值。需要注意的是,有一列会给出校正后的每个基因的p值。默认情况下,通过p.adjust函数中的Benjamini-Hochberg方法用于校正p值,如下所示:
|
|
在这里,我们比较一下使用limma分析的结果与以前得到的火山图结果。注意,红点现在都位于-log10(p.value)=2之下。此外,表示实际差异的蓝点的p值比以前更高,如下所示:
|
|

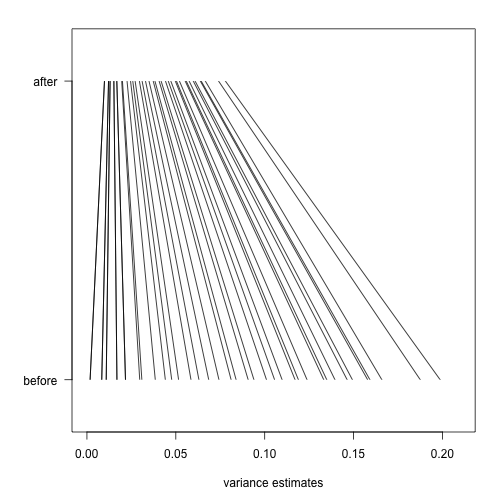
最后我们来构建一张图,用于展示limma是如何将方差估计缩小到一个共同值(common value)的,这能消除由于方差估计过低而导致的假阳性。
在这里,我们为不同的方差估计值设计40个小区间(bin),每个区间中落入一个基因。我们会删除那些不含有任何基因的区间,原文如下:
Here we pick, for each of 40 bins of different variance estimates, a single gene which falls in that bin. We remove bins which do not have any such genes.
|
|
现在绘制一条直线,这个直线展示了那些基因最初的估计方差到运行了limma之后的估计方差,如下所示:
|
|
关于limma校正的一些层级模型资料,可以参考以前的笔记《DALS019-统计模型2-贝叶斯分布与层次分析》。
参考资料
- Using limma for microarray analysis
- Smyth GK, “Linear models and empirical bayes methods for assessing differential expression in microarray experiments”. Stat Appl Genet Mol Biol. 2004 http://www.ncbi.nlm.nih.gov/pubmed/16646809